:: _cmdline:
cosolvkit command line interface
The CosolvKit command line interface is the easiest method to create and simulate a cosolvent system. If this is your first time learning about CosolvKit, take a look at the page Get started.
CosolvKit inputs
The script create_cosolvent_system.py provide all the necessary tools to build a cosolvent system and optionally run an MD simulation with standard setup. The main entry point of the script is the file config.json where all the necessary flags and command line options are specified. A template for the config.json can be found in cosolvkit/data/config.json. Cosolvents and forcefields templates can be found in the folder cosolvkit/data/ as well.
Argument |
Type |
Description |
Default value |
OPENMM |
AMBER |
GROMACS |
CHARMM |
|---|---|---|---|---|---|---|---|
cosolvents |
string |
Path to the json file containing the cosolvents to add to the system. |
no default |
||||
forcefields |
string |
Path to the json file containing the forcefields to use. |
no default |
||||
md_format |
string |
Format to use for the MD simulations and topology files. Supported formats: [OPENMM, AMBER, GROMACS, CHARMM] |
no default |
||||
receptor |
boolean |
Boolean describing if the receptor is present or not. |
no default |
||||
protein_path |
string |
If receptor is true this should be the path to the protein structure. |
no default |
||||
clean_protein |
boolean |
Flag indicating if cleaning the protein with PDBFixer |
TRUE |
||||
keep_heterogens |
boolean |
Flag indicating if keeping the heterogen atoms while cleaning the protein. Waters will be always kept. |
FALSE |
||||
variants |
dictionary |
Dictionary of residues for which a variant is requested (different protonation state) in the form {“chain_id:res_id”:”protonation_state”}, None for the rest of the residues. |
empty dictionary |
||||
add_repulsive |
boolean |
Flag indicating if adding repulsive forces between certain residues or not. |
FALSE |
✔️ |
✖️ |
✖️ |
✖️ |
repulsive_resiudes |
list |
List of residues for which applying the repulsive forces. |
empty list |
✔️ |
✖️ |
✖️ |
✖️ |
epsilon |
float |
Depth of the potential well in kcal/mol |
0.01 kcal/mol |
✔️ |
✖️ |
✖️ |
✖️ |
sigma |
float |
inter-particle distance in Angstrom |
10.0 Angstrom |
✔️ |
✖️ |
✖️ |
✖️ |
solvent_smiles |
string |
Smiles string of the solvent to use. |
H2O |
||||
solvent_copies |
integer |
If specified, the box won’t be filled up with solvent, but will have the exact number of solvent molecules specified. |
no default |
||||
membrane |
boolean |
Flag indicating if the system has membranes or not. |
FALSE |
||||
lipid_type |
string |
If membrane is TRUE specify the lipid to use. Supported lipids: [“POPC”, “POPE”, “DLPC”, “DLPE”, “DMPC”, “DOPC”, “DPPC”] |
“POPC” |
||||
lipid_patch_path |
string |
If the lipid required is not in the available, it is possible to pass a pre-equilibrated patch of the lipid of interest. |
no default |
||||
cosolvent_placement |
integer |
Integer deciding on which side of the membrane to place the cosolvents. Available options: [0 -> no preference, 1 -> outside, -1 -> inside] |
0 |
||||
waters_to_keep |
list |
List of indices of waters of interest in a membrane system. |
no default |
||||
radius |
float |
If no receptor, the radius is necessary to set the size of the simulation box. |
no default |
||||
output |
string |
Path to where save the results. |
no default |
||||
run_cosolvent_system |
boolean |
Flag indicating if running creating the system or not. |
TRUE |
||||
run_md |
boolean |
Flag indicating if running the md simulation after creating the system or not. |
FALSE |
✔️ |
✖️ |
✖️ |
✖️ |
CosolvKit can be run with and without protein (receptor), variants for the protonation states can be specified in the form of a python dictionary and custom repulsive forces can be specified between specific molecules in the system. The flag run_cosolvent_system decides if a new cosolvent system will be created, while the run_md flag takes care of running the MD simulation using the standard protocol provided by CosolvKit and generate trajectories (please note that this task is resources and time intensive depending on the hardware).
Post-processing pipeline
The script post_simulaiton_processing.py takes care of analysing the MD simulation trajectories and produces RDF plots as well as densities analysis as PyMol sessions. To access help message type:
$ post_simulation_processing.py --help
The script is based on the Report class and the following functions:
log_file: is the statistics.csv or whatever log_file produced during the simulation. At least Volume, Temperature and Pot_e should be reported on this log file.
traj_file: trajectory file
top_file: topology file
cosolvents_file: json file describing the cosolvents
- generate_report():
- out_path: where to save the results. 3 folders will be created:
- report
autocorrelation
rdf
- generate_density_maps():
out_path: where to save the results.
analysis_selection_string: selection string of cosolvents you want to analyse. This follows MDAnalysis selection strings style. If no selection string, one density file for each cosolvent will be created.
- generate_pymol_report()
selection_string: important residues to select and show in the PyMol session.
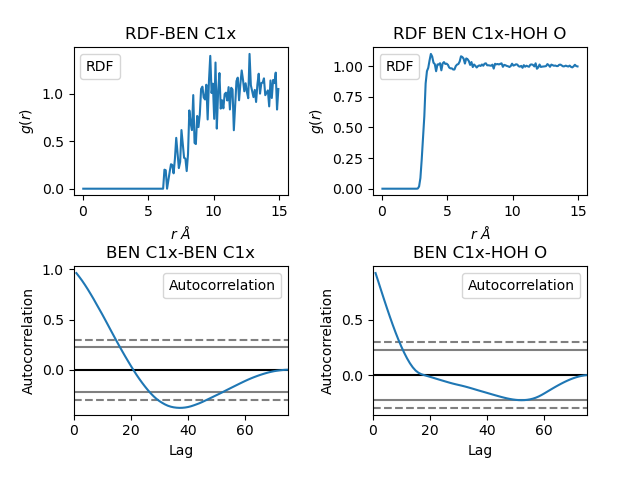
Example of an RDF plot generated with the post-processing pipeline.
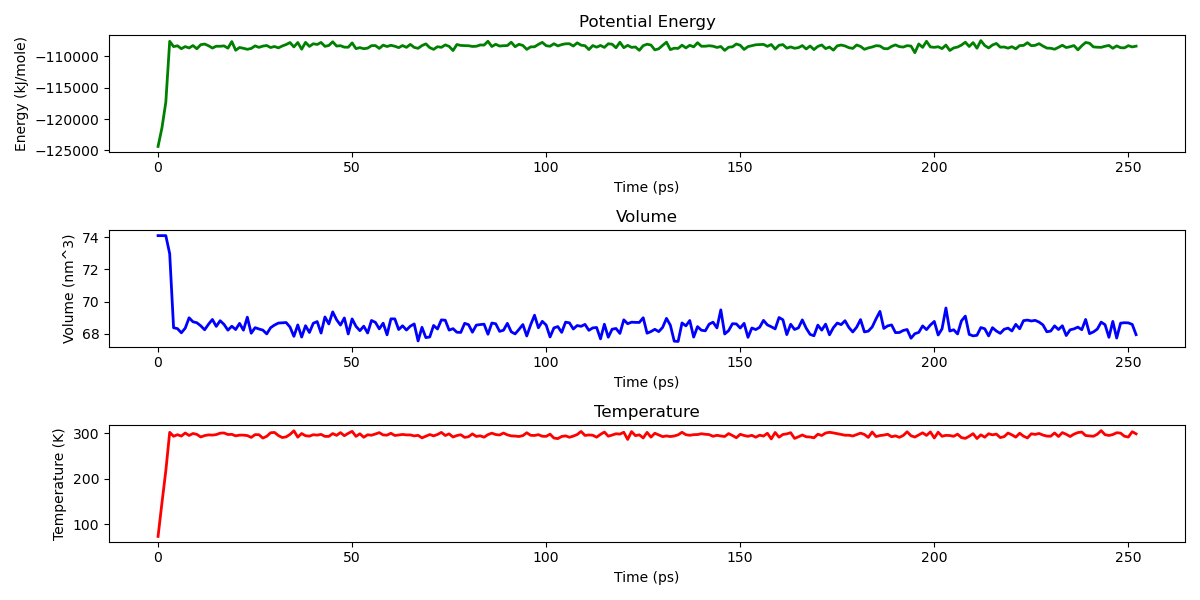
Example of a statistics plot generated with the post-processing pipeline.
Outputs
CosolvKit generates topology and positions files that will be used to run the MD simulation, the output format is decided by the field md_format in the config file.
Access help message
$ create_cosolvent_system.py --help